국제
사렙타, '실패보완' AAV DMD 2상 "추가 데이터 발표"
바이오스펙테이터 차대근 기자
AAV 기반 유전자치료제 후보물질 'SRP-9001'..자연사 코호트(natural history cohort)와 비교시 4~7세 DMD 환자 운동능력 평가지표(NSAA) 유의미한 개선 확인

사렙타 테라퓨틱스(Sarepta Therapeutics)가 뒤센근이영양증(Duchenne Muscular Dystrophy, DMD) 유전자치료제 후보물질 'SRP-9001(rAAVrh74.MHCK7.micro-dystrophin)' 임상2상 실패 후 4~7세 환자들의 운동능력 평가지표(NSAA) 개선을 증명할 추가 데이터 분석결과를 내놨다.
사렙타는 올해초 발표했던 임상2상 실패의 원인으로 6~7세 위약군이 임상시작(baseline)부터 증상이 약했던 것이 문제라고 판단해 자연사 코호트(natural history cohort)와 비교분석한 결과를 제시했다. 자연사 코호트는 특정 질병이 있는 사람들을 시간에 따라 추적 관찰하는 후향적 연구다.
사렙타는 지난 11일(현지시간) DMD 환자를 대상으로 진행한 SRP-9001의 임상2상과 1/2상에서 SRP-9001를 투여받은 4~7세 환자들이 운동능력 평가 지표인 NSAA(North Star Ambulatory Assessment) 점수를 개선한 결과를 보였다고 밝혔다.
SRP-9001의 임상2상에는 4~7세 DMD 환자 41명이 참여했다(NCT03769116). 환자들 중 무작위로 선정된 20명은 SRP-9001, 21명은 위약을 투여받았다. 1차 종결점은 12주차에 웨스턴블롯으로 측정한 마이크로 디스트로핀(Micro-dystrophin)의 변화와 48주차에 NSAA 점수 변화였다.
사렙타가 지난 1월 발표한 임상2상 탑라인 결과에 따르면, SRP-9001을 투여받은 환자들은 투여후 12주차에 평균 28.1%의 마이크로 디스트로핀 발현 증가를 보이며 1차 종결점을 충족했다(p<0.0001). 반면 48주차에 평가한 NSAA 점수는 위약 대비 수치적으로는 증가했으나 통계적으로 유의미하지 않았다(p=0.37).
사렙타는 연령별 하위그룹 분석에서 4~5세 코호트의 치료군은 NSAA 점수에서 4.3점의 개선을 보이며 위약군이 보인 1.9점 개선 대비 유의미한 차이를 보였으나(p=0.0172), 6~7세 코호트에서는 임상시작 당시부터 치료군보다 위약군의 점수가 유의미하게 좋았다고 설명했다(p=0.0046). 6~7세 위약군 환자들의 증세가 치료군만큼 심각하지 않아 치료효과가 정확하게 비교되지 않았다는 설명이다.
사렙타는 12명의 6~7세 환자에게 SRP-9001을 투여하고 1년후 NSAA 점수를 자연사 코호트와 비교했다. SRP-9001 투여군은 자연사 코호트 대비 NSAA 점수가 2.9점 개선됐다(p=0.0129).
사렙타는 DMD 환자 대상으로 SRP-9001의 효능을 본 임상 1/2상의 4~7세 환자들에게서도 자연사 코호트 대비 NSAA 점수가 개선된 것을 확인했다. 임상1/2상에서 SRP-9001를 투여 받은 4~7세 환자 4명은 투여 3년후 자연사 코호트 대비 NSAA 점수가 8.6점 개선됐다(p<0.0001)(NCT03375164).
도우 인글램(Doug Ingram) 사렙타 회장 겸 대표는 “SRP-9001를 통해 77명의 환자가 치료받았으며, 현존하는 DMD 유전자치료제 중 가장 포괄적이고 장기간 연구된 데이터”라며 “여러 연구에서 나온 증거들은 SRP-9001이 높은 발현과 지속적인 효능을 나타내는 단일투여 유전자치료제 후보물질임을 보여준다”고 말했다.
DMD는 근육막 기능유지에 필요한 디스트로핀 유전자에 돌연변이가 생겨 근섬유 퇴행이 일어나는 질병이다. 일반적으로 영유아시기에 발병해 걷기, 계단 오르기 등과 같은 발달 장애를 일으킨다. 환자들은 시간이 지남에 따라 근력 약화가 퍼지면서 점차 일상적인 행동수행 능력들을 상실하며, 호흡곤란이나 심장기능 장애 등의 위험이 커지게 된다.
SRP-9001은 아데노연관바이러스(AAV) 벡터를 통해 마이크로 디스트로핀 유전자를 전달하는 유전자치료제 후보물질이다. 비임상 연구에서 근육세포에 높은 친화도를 보인 AAVrh74를 사용한다. 로슈(Roche)와 지난 2019년에 맺은 라이선스 계약에 따라 미국내에서는 사렙타가, 미국 외에서는 로슈가 SRP-9001의 상업화 권리를 가지고 있다.
한편, 사렙타는 지난 4일 DMD 환자를 대상으로 SRP-9001의 효능을 확인하는 임상3상을 시작한다고 발표했다. 사렙타는 임상3상 참여 환자들의 50% 이상은 4~5세이며, 엑손1~17 사이에 돌연변이가 있거나 엑손45를 완전히 포함하는 돌연변이(inclusive)를 가진 환자들은 제외된다고 설명했다. 환자들은 무작위로 SRP-9001 또는 위약을 투여받는다. 1차 종결점은 투여 52주차에 NSAA 점수 변화다.
또한 경쟁사인 화이자(Pfizer)는 지난달 AAV 벡터 기반 DMD 치료제 후보물질 임상3상에서 나타난 심각한 이상반응 때문에 특정 돌연변이를 가진 환자를 제외하도록 임상 프로토콜을 수정한 바 있다.
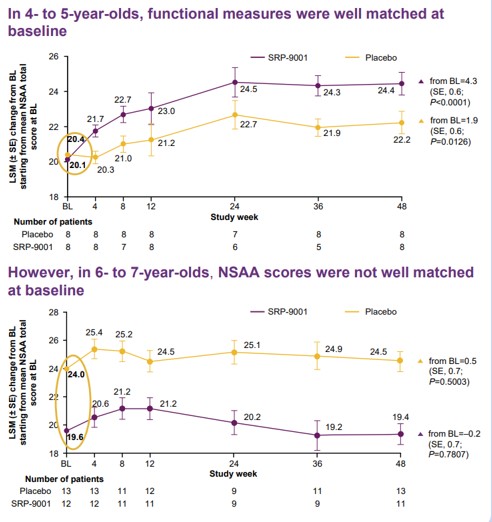
▲DMD 임상2상 연령별 NSAA 점수(사렙타 발표자료)










![[인사]대원제약, 2026년 정기 승진 인사](https://img.etoday.co.kr/crop/77/77/1956956.jpg)





