국제
크리스퍼-버텍스 "CRISPR 상업화 임상 유럽서 최초개시"
바이오스펙테이터 이은아 기자
ex-vivo 방식 'CTX001', 수혈 의존성 베타지중해빈혈 환자 12명 대상 임상1/2상 독일서 진행

제약사 주도 CRISPR 기반 치료제의 임상시험이 유럽에서 처음 개시된다. CRISPR 적용 임상은 2016년 중국에서 처음 시작됐으나 제약사가 주도한 상업화 임상시험은 이번이 처음이다.
3일 업계에 따르면 크리스퍼 테라퓨틱스(CRISPR Therapeutics)와 버텍스 파마슈티컬스(Vertex Pharmaceuticals)는 CRISPR 기반 치료제 후보물질 ‘CTX001'의 임상1/2상을 독일에서 지난달 27일 개시했다. 임상시험은 수혈 의존성 베타지중해빈혈(Transfusion-dependent β-thalassemia, TDT) 환자 12명을 대상으로 한다.
CTX001은 ex-vivo 방식의 유전자치료법이다. CRISPR 기술을 이용해 빈혈 환자의 조혈모세포의 베타글로빈 유전자 돌연변이를 교정함으로써 태아 헤모글로빈 수준을 증가시켜 다시 환자에게 주입한다. 현재 CTX001은 베타지중해빈혈(beta thalassemia)과 겸상적혈구 빈혈(sickle cell disease) 치료제로 개발하고 있다.
이번 유럽임상의 대상인 수혈 의존성 베타지중해성 빈혈(TDT)은 베타글로빈의 돌연변이로 비정상적인 적혈구를 생산해 헤모글로빈이 결핍되면서 심한 빈혈을 일으키는 유전질환이다. 적혈구가 산소를 제대로 공급하지 못하기 때문에 환자들은 15~20일 주기로 지속적인 수혈을 받아야한다. TDT는 수혈 외에는 마땅한 치료법이 없어 현재 다수의 유전자치료법이 개발되고 있는 상황이다.
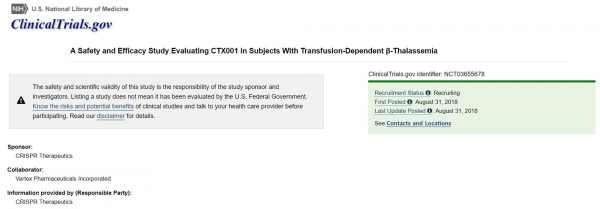
▲CRISPR Therapeutics의 'CTX001' 임상연구가 https://clinicaltrials.gov/에 등록됐다. 임상시험 코드( NCT03655678)
clinicaltrials.gov에 등록된 내용에 따르면, 크리스퍼와 버텍스는 18~35세 non-β0/β0 타입형 TDT 환자를 대상으로 단일군(single-arm), 오픈라벨, 단일용량으로 CTX001 임상1/2상을 지난 27일 착수했다. CTX001로 유전자 변형한 CD34+ 인간 조혈모세포 및 전구세포(hHSPCS)의 안전성과 효능을 평가한다.
1차 평가지수로 CTX001 투여 후 최소 6개월간 수혈 감소정도를 평가한다. 2차 평가항목은 6개월째 수혈 독립성, 이식 후 생착(engraftment) 비율, 호중구 및 혈소판 생착에 걸리는 시간, 부작용 심각성 및 빈도, 이식 관련 사망률 등이다. 이번임상은 독일 Regensburg의 한 병원에서 수행되며, 2021년 2월 1차 결과를 발표하고 2022년 임상을 종료한다는 목표다.
크리스퍼 테라퓨틱스와 버텍스는 2015년부터 CRISPR 기반 치료제 개발을 위한 공동연구를 진행해왔다. 당시 버텍스는 크리스퍼에 현금 7500만달러와 3000만달러 규모의 지분투자를 했다. 계약에 따라 버텍스는 최대 6개 후보물질의 기술이전에 대한 권리를 확보했으며, CTX001은 지난 12월 독점적 권리를 확보한 첫번째 자산이다. 크리스퍼와 버텍스는 공동개발을 추진하며 상업화에 따른 이윤을 공동 분배한다.
한편 크리스퍼와 버텍스는 지난 5월 FDA로부터 'CTX001'의 겸상적혈구빈혈 임상1/2상 신청에서 임상보류 통지를 받은 바 있다.
에디타스 메디슨(Editas Medicine)도 in vivo 방식의 ‘EDIT-101' 임상시험이 다소 지연되고 있다. 회사 측은 "제조 문제로 다소 늦어진 임상은 오는 10월 FDA에 임상시험신청(IND)을 제출할 계획"이라고 밝혔다. EDIT-101은 선천성 망막질환인 레베르선천성흑내장(Laber's congenital amaurosis, LCA) 치료를 목표로 개발 중이다.











![[인사]대원제약, 2026년 정기 승진 인사](https://img.etoday.co.kr/crop/77/77/1956956.jpg)




