오피니언
[남궁석의 신약연구史]'LDL 리셉터' 실체 밝혀진 과정
남궁석 SLMS(Secret Lab of Mad Scientist) 대표
스타틴을 찾아서③ LDL과 콜레스테롤의 분자적인 조절기전 밝힌 핵심 인물 조셉 L. 골드슈타인과 마이클 브라운
이번 연재분에서는 우리가 현재 알고 있는 LDL과 콜레스테롤의 분자적인 조절기전을 규명하는데 큰 역할을 한 두 과학자, 즉 조셉 L. 골드슈타인(Joseph L Goldstein, 1940-)과 마이클 브라운(Micheal Brown, 1941-)의 발견을 중심으로 하여 LDL 리셉터(LDL Receptor)와 콜레스테롤의 조절 기작을 알아보도록 하자.
유전성 과다콜레스테롤증(Familial hypercholesterolemia, FH)
이전 연재에서 기술한 것처럼 유전성 과다콜레스테롤증은 1939년부터 보고되어 있었으나, 과연 어떤 기전에 의해서 이러한 이상이 초래되는지는 1970년대까지 전혀 알려지지 않았다. 다만 1964년 레바논의 연구자인 아베디스 차카듀리안(Avedis K. Khachadurian)은 유전성 과다콜레스테롤증을 유발하는 유전변이에는 좀 더 증상이 심한 동형접합(homozygote)과 증상이 상대적으로 덜한 이형접합(heterozygote)의 두 가지 형태가 있다는 것을 보고했다[1]. 유전성 과다콜레스테롤증을 유발하는 유전자 중 한 카피에서만 결함이 있는 이형접합의 경우 약 500명에 1명 정도 나타나는 비교적 흔한 유전변이이고, 출생시부터 혈중 LDL 농도가 정상인에 비해서 2배 정도 높다. 60세 이전에 심근경색을 일으키는 환자 중 약 5%가 유전성 과다콜레스테롤증의 이형접합을 가지고 있다[2]. 반면 두 카피의 유전자에 모두 결함을 가지고 있는 동형접합 유전성 과다톨레스테롤증은 100만명 중 1명 정도가 발견되는 극히 희귀하며, 정상인에 비해서 혈중 LDL 농도가 6-10 배 정도 높으며, 어린 시절부터 죽상경화나 심근경색이 나타나는 경우가 많다. 1964년 3세 소년이었던 존 데스포타(John Despota)라는 환자는 전형적인 유전성 과다콜레스테롤 환자였고 그의 표현형은 동형접합 환자와 비슷하게 나타났다. 바로 이 환자가 가지고 있던 유전 변이가 LDL이 세포 내로 흡수되고, 세포 내의 콜레스테롤 합성을 조절하는지를 규명하는데 결정적인 역할을 했다.
LDL 리셉터
원래 내과의로 훈련되었으나, 전공의 이후에 NIH에서 포스트닥으로 효소 생화학 연구 경험을 쌓았던 골드슈타인과 브라운은 1972년 댈러스의 텍사스대학 의과대학교(UT Southwestern Medical School)에 임용되면서 콜레스테롤 관련 연구에 뛰어들었다. 이들은 기존에 실험동물 중심으로 수행되던 콜레스테롤 대사 관련 연구를 인간을 대상으로 수행하기로 했다. 그렇다면 어떤 모델 시스템을 사용해야 하는가? 이미 그당시에도 콜레스테롤의 생합성은 주로 간이나 소장에서 일어난다는 것이 알려져 있었다. 그러나 인간의 간이나 소장을 이용하여 콜레스테롤 연구를 하는 것은 현실적으로 불가능하였다. 이들은 유전성 과다콜레스테롤환자 유래의 피부세포를 모델 시스템으로 하여 연구를 수행하기로 하였다.
이들은 유전성 과다콜레스테롤 환자의 경우 1960년대에 규명된 콜레스테롤 생합성 경로의 주요 속도조절 단계인 HMG-CoA 환원효소(HMG-CoA reductase)의 활성이 정상인과 다를 것이라는 가설을 세웠다. 이를 입증하기 위하여 이들은 일단 정상 세포의 HMG-CoA 활성효소가 혈액중의 LDL 농도와 어떤 관계가 있는지를 관찰했다. LDL을 포함하고 있는 일반적인 혈청에서 배양될 때 피부세포에서의 HMG-CoA 환원효소의 활성은 매우 낮았다. 그러나 혈청에서 LDL을 제거하자 피부에서 HMG-CoA 환원효소의 활성은 급격하게 올라갔고, 배양액 중에 LDL을 다시 첨가하자 HMG-CoA 환원효소의 활성은 다시 내려갔다. 이것은 혈액 중에서 LDL의 형태로 콜레스테롤이 얼마나 많이 존재하는 것을 세포가 감지하여 콜레스테롤 합성을 조절한다는 의미이다. 즉 혈액 중에 LDL 콜레스테롤이 존재하여 이를 흡수할 수 있다면 세포에서 콜레스테롤 합성을 하지 않지만, LDL 콜레스테롤이 없어지면 콜레스테롤 합성을 하게 된다는 것이다. 그런데 동형접합 FH 환자로부터 얻은 세포의 경우 전혀 다르게 행동했다. 즉, 이 세포에서는 혈액 중의 LDL이 있는 것과 상관없이 정상세포에 비해서 50-100배 이상 높은 양의 HMG-CoA 환원효소가 검출되었다. 즉, 혈액 중의 콜레스테롤과 상관없이 이 세포에서는 콜레스테롤을 생산한다는 것이다[3]
그렇다면 왜 이러한 일이 일어날까? 가장 간단한 해석은 콜레스테롤이 존재하면 그 활성이 줄어드는 HMG-CoA 활성효소에 돌연변이가 생겨서, 콜레스테롤 존재하에서도 활성이 유지된다는 것이다. 그러나 이들은 LDL 없이 에탄올과 콜레스테롤을 섞어서 세포에 넣을 수 있는 방법을 통해서 실험해 본 결과, LDL 없이 세포에 콜레스테롤을 넣으면, FH 환자의 세포에서도 HMG-CoA 환원효소의 활성은 줄어든다는 것을 확인하였다. 즉 이들이 발견한 현상은 콜레스테롤을 합성하는 HMG-CoA 환원효소에서의 문제가 아닌 세포가 어떻게 LDL을 흡수하는지에 관련된다는 것이다.[4].
이들은 1974년 LDL을 동위원소(I125)로 표지한 후, 이렇게 동위원소로 표지된 LDL이 정상세포와 이전에 발견된 FH 동형접합 환자인 존 데스포라 유래의 세포에서 어떻게 흡수되는지를 관찰하였다. 그 결과 정상 세포에는 특이적으로 LDL을 결합하는 부위가 존재하지만, FH 환자의 경우에는 그렇지 않아 LDL이 세포 내로 흡수되지 않고 세포막에 퍼져 있음이 발견되었다[5]. 정상 세포에서는 동위원소로 표시된 LDL이 흡수된 이후, LDL은 분해되어 콜레스테롤이 세포 내로 흡수되지만, 환자 세포에서는 LDL 자체가 흡수되는 것이 불가능했기 때문이다. 결국 FH 환자에서 문제가 있던 유전자는 LDL 을 세포로 흡수하는, LDL 리셉터(LDL Receptor) 유전자였다. 즉 LDL 입자가 세포 표면에 존재하는 LDL 리셉터에 결합하고, 이에 인해 세포 내부로 흡수되는 ‘리셉터 기반 엔도사이토시스’(Receptor-mediated Endocytosis)의 존재가 최초로 발견되게 된 셈이다.
![▲그림 1. LDL 리셉터 발견의 주역인 마이클 브라운 (좌) 와 조셉 골드슈타인 (우). 이들은 유전성 고콜레스테롤증 환자의 피부세포를 모델 시스템으로 이용하여 혈액 중의 LDL의 흡수가 세포내의 콜레스테롤 생합성과 직접적으로 관련있다는 것을 규명하였다. 정상세포의 경우 혈액 중에서 LDL을 제거하면 콜레스테롤 생합성의 핵심 단계인 HMG 환원효소의 수준이 점차 증가하나, 유전성 고콜레스테롤증 환자 유래의 세포에서는 HMG 환원효소가 LDL의 존재 여부와 관계없이 항상 높게 나타난다. 유전성 고콜레스테롤증 환자는 LDL을 세포로 흡수할 수 없으며,따라서 LDL를 세포 내에서 분해하여 콜레스테롤을 흡수할 수 없다. 정상 세포는 혈액 중의 LDL을 흡수하여 콜레스테롤을 흡수하고, 세포 내의 콜레스테롤합성은 억제되는 반면, 유전성 고콜레스테롤증 환자에서는 혈액 중의 LDL을 흡수할 수도 없고, 세포 내에서의 HMG 환원효소는 계속 활성화되어 계속 콜레스테롤을 합성하기 때문에 LDL 유래의 콜레스테롤 및 세포 유래의 콜레스테롤 합성 모두 증가하게 된다[6].](http://img.etoday.co.kr/pto_db/2018/10/20181017014539_1259951_692_733.JPG)
▲그림 1. LDL 리셉터 발견의 주역인 마이클 브라운 (좌) 와 조셉 골드슈타인 (우). 이들은 유전성 고콜레스테롤증 환자의 피부세포를 모델 시스템으로 이용하여 혈액 중의 LDL의 흡수가 세포내의 콜레스테롤 생합성과 직접적으로 관련있다는 것을 규명하였다. 정상세포의 경우 혈액 중에서 LDL을 제거하면 콜레스테롤 생합성의 핵심 단계인 HMG 환원효소의 수준이 점차 증가하나, 유전성 고콜레스테롤증 환자 유래의 세포에서는 HMG 환원효소가 LDL의 존재 여부와 관계없이 항상 높게 나타난다. 유전성 고콜레스테롤증 환자는 LDL을 세포로 흡수할 수 없으며,따라서 LDL를 세포 내에서 분해하여 콜레스테롤을 흡수할 수 없다. 정상 세포는 혈액 중의 LDL을 흡수하여 콜레스테롤을 흡수하고, 세포 내의 콜레스테롤합성은 억제되는 반면, 유전성 고콜레스테롤증 환자에서는 혈액 중의 LDL을 흡수할 수도 없고, 세포 내에서의 HMG 환원효소는 계속 활성화되어 계속 콜레스테롤을 합성하기 때문에 LDL 유래의 콜레스테롤 및 세포 유래의 콜레스테롤 합성 모두 증가하게 된다[6].
LDL 리셉터 유전자의 발견과 이의 조절
이제 LDL 리셉터의 실체가 알려지는 과정을 알아보도록 하자. 1982년 브라운과 골드슈타인 연구실에서는 소의 부신(副腎, adrenal glands)에서 동위원소로 표지한 LDL을 결합하는 단백질인 LDL 리셉터 단백질을 정제하고, 이것이 분자량이 164,000 에 달하는 막단백질이라는 것을 확인하였다[7]. 1984년 단백질의 아미노산 정보를 이용하여 제작된 유전자 탐침(Probe)을 이용하여 LDL 리셉터의 cDNA가 클로닝되었고, LDL 리셉터의 N말단에는 LDL입자를 결합하는 리간드 결합 도메인, 가운데는 EGF와 유사한 서열, 그리고 생체막을 가로지르는 도메인이 있으며 세포질에 노출되어 있는 C말단의 영역에는 리셉터를 엔도사이토시스에 의해서 세포로 함입되는 영역으로 유도하는 부분이 있다는 것을 확인할 수 있었다[8]
![▲그림 2. LDL 리셉터 유전자는 N말단에 LDL이 붙는 LA 반복부분과 가운데 부분의 EGF 유사 영역, 그리고 생체막을 통과하는 영역과 약 50아미노산 가량으로 구성된 세포질 내 부분으로 구성되어 있다. LDL가 세포 내로 흡수되지 못해서 유전성 고콜레스테롤혈증을 앓게 된 존 데스포라의 LDL 리셉터에는 세포질 내 부분에 돌연변이가 존재하여 (점으로 표시된 부분) LDL 리셉터가 엔도사이토시스로 세포 내로 이동하지 못하게 되었다. LDL 은 세포 표면에서 LDL 리셉터의 LA 반복부위에 붙어서 흡수되고, 세포 내의 엔도솜 (endosome)에서 pH가 올라가면 LDL 리셉터와 떨어져서 콜레스테롤을 방출하게 된다[10].](http://img.etoday.co.kr/pto_db/2018/10/700/20181017014557_1259953_898_282.JPG)
▲그림 2. LDL 리셉터 유전자는 N말단에 LDL이 붙는 LA 반복부분과 가운데 부분의 EGF 유사 영역, 그리고 생체막을 통과하는 영역과 약 50아미노산 가량으로 구성된 세포질 내 부분으로 구성되어 있다. LDL가 세포 내로 흡수되지 못해서 유전성 고콜레스테롤혈증을 앓게 된 존 데스포라의 LDL 리셉터에는 세포질 내 부분에 돌연변이가 존재하여 (점으로 표시된 부분) LDL 리셉터가 엔도사이토시스로 세포 내로 이동하지 못하게 되었다. LDL 은 세포 표면에서 LDL 리셉터의 LA 반복부위에 붙어서 흡수되고, 세포 내의 엔도솜 (endosome)에서 pH가 올라가면 LDL 리셉터와 떨어져서 콜레스테롤을 방출하게 된다[10].
그 이후 LDL 자체가 세포 내에 흡수되지 않고 표면에 붙어있던 것이 관찰되던 환자인 존 데스포라의 경우 두 카피의 유전자 중 하나는 단백질 자체가 만들어지지 않는 돌연변이를 가지고 있었고, 다른 카피의 경우 C말단 영역에서 돌연변이가 있었고, 이 돌연변이(정상인에서는 타이로신인 807번째 아미노산이 시스테인으로 바뀐)의 경우 리셉터의 정상적인 엔도사이토시스를 막는 것이 확인되었다[9].
이러한 돌연변이를 포함하여 약 1000종 이상의 LDL 리셉터의 돌연변이가 LDL 리셉터의 기능을 억제하고, 이에 따라서 LDL의 정상적인 세포에서의 흡수를 억제하고, 이에 따라서 세포 내에서의 콜레스테롤의 생합성이 조절되지 않고 계속 유지된다는 것이 확인되었다. 이러한 공로에 의해서 골드스테인과 브라운은 1985년 노벨 생리학상을 수상하였다. 그러나 콜레스테롤에 의한 LDL 리셉터의 조절에 관해서는 이들이 노벨상을 받을 때까지 풀리지 않았던 문제가 있었다.
LDL 리셉터에 의해서 세포 내로 유입된 LDL이 세포 내에서의 콜레스테롤의 생합성을 억제하는 것 뿐만 아니라, 세포 내에 충분한 콜레스테롤이 유입되면 LDL 리셉터가 만들어지는 것도 억제한다[1-]즉 세포 내에 콜레스테롤이 충분히 존재한다면 굳이 세포가 콜레스테롤을 흡수할 필요가 없고, 불필요한 LDL 리셉터가 만들어지는 것도 억제될 필요가 있기 때문이다. 그렇다면 세포 내의 LDL 리셉터가 만들어지는 것은 어떻게 조절되는가? LDL 리셉터가 만들어지는 것이 조절되기 위해서는 세포 내의 콜레스테롤에 의해서 LDL 리셉터의 전사가 조절되어야 한다. 그런데 콜레스테롤은 세포막에 있으며, 전사가 조절되기 위해서는 그 신호가 핵까지 전달되어야 된다. 어떻게 세포 내의 콜레스테롤 수준에 따라서 LDL 리셉터가 조절될까?
1993년 LDL 리셉터의 유전자 발현을 조절하는 중요한 조절인자인 SREBP-1(Sterol regulatory element binding protein-1)이라는 단백질이 발견되었다[11]. 이 단백질은 ER-골지의 세포막에 존재하는 단백질로써, 콜레스테롤 결핍시에는 골지로 이동한 후 단백질 분해효소인 S1P/S2P에 의해서 절단되어, LDL 리셉터의 유전자 발현을 활성화하는 전사인자 부분이 핵으로 이동하고, LDL 리셉터의 유전자 발현을 활성화하여 더 많은 LDL 리셉터를 만들게 된다.
그러나 세포 내 콜레스테롤의 농도가 증가하여 ER 내의 세포막의 콜레스테롤 농도가 5% 이상이 되면, SREBP는 더이상 단백질이 잘려지는 장소인 골지로 이동하지 못하게 되고 따라서 핵으로 이동하여 LDL 리셉터의 유전자 발현을 활성화하지 못하게 된다. 결국 세포 내의 콜레스테롤이 많아지면 더 이상 LDL 리셉터를 만들지 못하게 되는 셈이다[12].
이렇게 콜레스테롤 수준에 의해서 LDL 리셉터의 양이 조절되는 것은 다음 회에 설명할 콜레스테롤 합성 저해제인 스타틴(Statin)에 의해서 혈중 LDL 수준이 낮아지는것과 직접적인 관계가 있다. 즉 스타틴에 의해서 세포 내에서 콜레스테롤 합성이 줄어들게 되면, 세포에서는 더 많은 양의 LDL 리셉터를 만들게 되고, 더 많이 만들어진 LDL 리셉터는 혈관 중의 LDL과 결합하여 더 많은 LDL을 세포 내로 흡수시키게 하고, 따라서 혈중의 LDL 농도를 낮추게 되는 것이다.
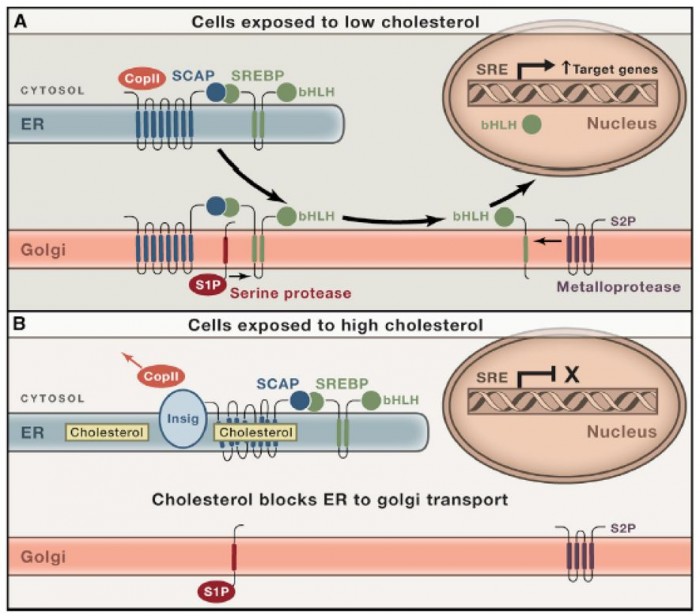
▲그림 3 : SREBP에 의한 LDL 리셉터 유전자 발현의 조절. 콜레스테롤이 없는 상태에서는 LDL 리셉터 등의 유전자 발현을 촉진하는 전사인자인 SREBP는 ER에서 골지로 이동하고, 골지체에서 단백질 분해효소인 S1P/S2P에 의해서 전사를 촉진하는 부분만 분리되어 핵으로 이동하여 LDL 리셉터의 전사를 촉진하는 유전자들을 발현시킨다. 그러나 콜레스테롤이 ER의 생체막에 5% 이상 존재할 경우 SREBP는 골지체로 수송되지 못하고 남아있게 되고, 따라서 핵으로 이동하여 전사를 촉진할 수 없기 때문에 LDL 리셉터의 발현은 억제된다. 이렇게 세포 내의 콜레스테롤 수준에 따라서 LDL 리셉터가 만들어지는 양이 조절된다.
이렇게 1970년대부터 진행된 LDL 리셉터에 대한 연구는 LDL을 세포 내로 흡수하는 과정과 L세포 내에서 콜레스테롤을 합성하는 과정은 매우 밀접한 관련이 있으며, 세포 내의 콜레스테롤의 합성을 조절함으로써 혈액 중의 LDL 역시 LDL 리셉터를 통하여 흡수하여 조절할 수 있다는 이론적 근거를 제시하게 되었다. 브라운과 골드스타인의 연구는 단지 콜레스테롤과 동맥경화증과의 관계를 넘어서 세포 내에 물질이 흡수되는 과정인 리셉터 의존성 엔도사이토시스의 기전을 최초로 제시한 것으로도 의미가 크다 하겠다.
다음은 드디어 콜레스테롤 생합성 경로를 조절하여 혈중 LDL 농도를 조절하는 약물인 스타틴(Statin)의 개발과정을 알아보도록 한다.
참고문헌
1, Khachadurian, A. K. (1964). The inheritance of essential familial hypercholesterolemia. The American journal of medicine, 37(3), 402-407.
2. Goldstein, J. L., & Brown, M. S. (2009). The LDL receptor. Arteriosclerosis, thrombosis, and vascular biology, 29(4), 431-438
3. Goldstein, J. L., & Brown, M. S. (1973). Familial hypercholesterolemia: identification of a defect in the regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity associated with overproduction of cholesterol. Proceedings of the National Academy of Sciences, 70(10), 2804-2808;Brown, M. S., Dana, S. E., & Goldstein, J. L. (1974). Regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity in cultured human fibroblasts Comparison of cells from a normal subject and from a patient with homozygous familial hypercholesterolemia. Journal of Biological Chemistry, 249(3), 789-796.
4. Goldstein, J. L., & Brown, M. S. (1974). Binding and Degradation of Low Density Lipoproteins by Cultured Human Fibroblasts Comparison Of Cells From A Normal Subject And From A Patient With Homozygous Familial Hypercholesterolemia. Journal of Biological Chemistry, 249(16), 5153-5162.
5. Anderson, R. G., Goldstein, J. L., & Brown, M. S. (1977). A mutation that impairs the ability of lipoprotein receptors to localise in coated pits on the cell surface of human fibroblasts. Nature, 270(5639), 695.
6. Goldstein, J. L., & Brown, M. S. (2015). A century of cholesterol and coronaries: from plaques to genes to statins. Cell, 161(1), 161-172.
7. Schneider, W. J., Beisiegel, U., Goldstein, J. L., & Brown, M. S. (1982). Purification of the low density lipoprotein receptor, an acidic glycoprotein of 164,000 molecular weight. Journal of Biological Chemistry, 257(5), 2664-2673.
8. Yamamoto, T., Davis, C. G., Brown, M. S., Schneider, W. J., Casey, M. L., Goldstein, J. L., & Russell, D. W. (1984). The human LDL receptor: a cysteine-rich protein with multiple Alu sequences in its mRNA. Cell, 39(1), 27-38.
9. Davis, C. G., Lehrman, M. A., Russell, D. W., Anderson, R. G., Brown, M. S., & Goldstein, J. L. (1986). The JD mutation in familial hypercholesterolemia: amino acid substitution in cytoplasmic domain impedes internalization of LDL receptors. Cell, 45(1), 15-24.
10. Jeon, H., & Blacklow, S. C. (2005). Structure and physiologic function of the low-density lipoprotein receptor. Annu. Rev. Biochem., 74, 535-562.
11. Brown, M., & Goldstein, J. (1986). A receptor-mediated pathway for cholesterol homeostasis. Science, 232(4746), 34–47. doi:10.1126/science.3513311
12. Yokoyama, C., Wang, X., Briggs, M. R., Admon, A., Wu, J., Hua, X., ... & Brown, M. S. (1993). SREBP-1, a basic-helix-loop-helix-leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell, 75(1), 187-197.













![[인사]일동홀딩스, 새 대표이사에 최규한 사장 선임](https://img.etoday.co.kr/crop/77/77/2313330.jpg)


![[인사]일동홀딩스, 새 대표이사에 최규한 사장 선임](https://img.etoday.co.kr/crop/74/74/2313330.jpg)


