피플
[남궁석의 신약연구史] 저온전자현미경(Cryo-EM) 혁명
남궁석 SLMS(Secret Lab of Mad Scientist) 대표
[단백질구조부터 신약까지⑦끝]전자현미경→저온전자현미경..네가티브 염색, 샘플 급속동결 등 테크닉 개발
지금까지 주로 단백질 결정학을 통해 생물학적으로 중요한 여러가지 단백질의 구조가 밝혀지는 과정을 알아보았다. 비록 여러가지 돌파구의 등장으로 이전보다 단백질 결정화가 용이해지긴 했지만, 여전히 단백질 결정화는 어려운 일이었고, 생물학적으로 중요한 많은 단백질 중 상당수는 그동안 개발된 여러가지 트릭을 총동원해도 결정화되지 않았다. 그렇다면 결정화되지 않는 단백질의 구조정보는 어떻게 얻을 수 있을까? 이번 연재에서는 2010년 이후 구조생물학의 주류 방법론이 된 '저온전자현미경(Cryo-EM)'이 어떤 과정을 통하여 단백질 구조규명에 이용되게 되었는지의 과정을 알아보도록 한다.
전자현미경의 개발
일단 전자현미경이 개발된 과정부터 알아보도록 하자. 17세기부터 이용된 광학현미경은 물체와 물체를 구분하는 해상력(Resolution Power)에 한계를 가지고 있었다. 1873년 독일의 광학자인 에른스트 아베는 광학현미경이 이용하는 가시광선의 파장길이의 절반이 실제 광학현미경에서 식별 가능한 물체의 한계선이라는 것을 발견하였다. 즉 400nm의 파장을 가지는 가시광선이라면 식별 가능한 가장 작은 물체는 200nm, 즉 0.2µ가 되는 셈이다. 물론 이 정도의 해상도는 직경이 대략 5-10nm인 단백질을 관찰하기는 턱없이 부족하고 직경 100nm 정도인 바이러스도 관찰하기 힘든 해상도였다.
이를 극복하기 위해서는 가시광선보다 훨씬 파장이 짧은 파동을 이용하는 현미경을 만들어야만 했다. 가시광선보다 훨씬 더 파장이 짧은 파동은 전자파다. 오늘날의 전자현미경, 보다 정확히 말하면 ‘투과 전자현미경(transmission electron microscope, TEM)'은 독일의 전기공학자 에른스트 루스카(Ernst Ruska)와 그의 지도교수인 막스 크놀(Max Knoll)에 의해 개발되었다. 투과 전자현미경은 빛 대신 높은 전압에서 나오는 전자를 이용한다는 것을 제외하고는 광학현미경과 원리가 비슷하다. 시료를 통과한 빛이 유리로 된 광학렌즈를 통과해 굴절하면서 상이 확대되는 것과 마찬가지로, 전자원에서 나온 전자는 전자석 코일을 통과하면서 굴절되며 상을 확대시키고, 확대된 시료의 정보를 담고있는 전자는 형광스크린이나 필름 혹은 이미지 센서에 의해서 감지되어 확대된 상을 표시하게 된다. 루스카는 1933년 1만2000배의 확대능을 가진 전자현미경을 제작했다. TEM의 해상도는 점점 개선되었고, 21세기에는 최대 50pm, 즉 0.5Å의 거리도 식별 가능할 정도의 고분해능을 얻을 수 있게 되었다.
그러나 전자현미경을 이용하여 단백질 등의 생체 고분자의 구조를 얻기에는여러가지 문제가 있었다. 일단 높은 에너지를 가진 전자선이 시료에 맞으면 시료가 손상되는 것이 문제였다. 그리고 TEM으로 시료를 촬영하기 위해서는 진공상태에서 촬영하여야 하는데, 단백질 등의 생물학 시료는 진공상태에서 건조되며 제대로 된 구조를 잃기 때문에 관찰하기 어렵다. 또한 생물시료는 탄소, 질소, 수소로 주로 구성되어 있는데 이들 원소는 전자를 산란하는 정도가 낮기 때문에 얻은 영상의 명암대비가 낮고, 고분해능을 유지하는 영상데이터를 얻기가 힘들었다. 이러한 여러가지 문제 때문에 전자현미경은 주로 세포내부의 구조 등을 알아내는데 사용되었으며, 단백질의 구조를 알아보는데의 응용은 상당히 더딘 편이었다.[1] 그러나 여러가지 테크닉이 개발되면서 단백질의 구조연구에도 서서히 전자현미경이 사용되기 시작했다.
전자현미경의 단백질 구조규명에 이용시작-네가티브 염색
일단 제일 문제는 단백질 분자가 전자를 잘 산란하지 않아서 영상이 선명하게 나오지 않는다는 문제였다. 이를 극복하기 위하여 네가티브 염색(Negative Staining)이라는 방법이 개발되었다. 이 방법은 단백질 등의 시료를 탄소로 코팅된 표면에 붙이고, 여기에 우라늄 아세트산(Uranium Acetate)과 같이 전자를 잘 산란하는 물질을 가하는 것이다. 단백질이 붙어있지 않는 표면에 우라늄 이온이 붙고 단백질이 붙어있지 않는 곳에는 붙지않는 성질을 이용하여 단백질 시료 바깥 부분을 검게 염색하여 단백질의 윤곽을 관찰하는 방법이다.[2] 비록 이 방법은 최대 해상도 20Å 정도로 단백질의 대략적인 윤곽밖에 식별할 수 없었지만, 이러한 저해상도의 데이터로도 생체고분자의 전체적인 모양에 대한 많은 정보를 얻을 수 있었다. 가령 리보좀의 대략적인 윤곽과 리보좀이 크기가 다른 2개의 서브유니트로 구성되어 있다는 정보,[3] 액틴이나 마이크로튜블 같은 세포골격 단백질의 대략적인 구조,[4] 프로테오솜의 구조[5]와 같이 우리가 현재 알고 있는 생체 고분자의 구조에 대한 많은 정보들이 1970-1980년대에 네가티브 염색을 통한 전자현미경 관찰로 얻어졌다. Cryo-EM이 일반화된 현재에도 네가티브 염색에 의한 단백질 관찰은 계속 유용하게 사용되고 있는데, 빠른 시간 안에 샘플의 상태를 관찰할 수 있다는 특징 때문에 Cryo-EM을 수행하기 전에 샘플의 상태를 확인하는 목적으로 많이 이용되고 있다.
물론 네가티브 염색은 염색 과정에서 단백질의 구조에 영향을 주고, 물이 존재하지 않는 진공상태이므로 생체고분자의 구조를 변형시킨다는 단점이 있다. 따라서 보다 정밀한 구조를 얻기 위해서는 다른 방식의 샘플 처리방법을 개발해야만 했다.
전자현미경을 이용한 2D 결정학
X선 회절이 아닌 전자현미경을 이용해도 원자 수준의 고해상도 단백질 구조 정보를 얻을 수 있다는 것이 처음 알려진 것은 박테리오로돕신(Bacteriorhodopsin)이라는 빛 에너지를 이용하여 ATP를 생산하는 고세균(Archaea) 유래의 단백질 구조 연구였다. 박테리오로돕신은 막단백질으로써, 고세균의 세포막에 결정 형태로 배열되어 있었다. 일반적인 X선 회절에 이용되는 결정은 평면과 두께 부분으로 단백질이 배열되는 3차원 결정이지만, 박테리오로돕신의 세포막의 결정은 평면으로만 배열되고 두께로는 생체막에 의해서 하나의 단백질만 배열되는 2차원 결정이었다. 이 연구를 수행하던 리차드 핸더슨(Richard Henderson)은 처음에는 박테리오로돕신의 2차원 결정을 분리하여 계면활성제로 용해한 후 3차원 결정을 만들려고 노력했지만, 이는 제대로 되지 않았다. 대신 전자현미경의 전자파를 2차원 결정에 쐬면 3차원 결정과 마찬가지로 회절을 한다는 것을 발견하였고, X선이 아닌 전자선의 회절 데이터를 이용하여 단백질 구조를 풀 수 있다는 것을 보였다. 1975년 해상도 7Å 정도의 구조를 처음 2차원 결정의 회절 데이터를 이용하여 얻었지만 이 때의 데이터는 해상도가 낮아 실제로 단백질의 아미노산이 어떻게 배열되어 있는지를 구분할 수 있는 수준은 아니었다.[6] 이후 약 10여년간의 노력을 통해 1990년 3Å대의 고해상도의 전자밀도를 얻었고, 이를 해석하여 박테리오로돕신의 구조를 풀 수 있었다.[7] 이 결과는 X선이 아닌 전자선을 이용해서 해상도 3Å급의 고해상도 구조를 얻을 수 있다는 것을 최초로 보여준 예가 되었다.
그러나 핸더슨이 사용한 방법은 여전히 2D 결정을 얻고 전자선 회절 데이터를 얻는 방법이었고, 대부분의 단백질들은 결정화가 어려웠기 때문에 이 방법은 폭넓게 적용되기는 어려였다. 결국 전자현미경을 이용하여 결정화되지 않은 단일 단백질 입자의 이미지를 얻고, 여기서부터 고해상도 구조를 얻는 방법이 개발되어야만 했다.
샘플 급속동결에 의한 Cryo-EM의 개발
1980년대 EMBL(European Molecular BIology Laboratory)의 연구자들은 그때까지 전자현미경 관찰의 근원적인 문제였던 ‘물'을 어떻게 처리하는지를 고민하고 있었다. 앞에서 언급했다시피 전자현미경 관찰은 진공상태에서 이루어지지만, 단백질 등의 생물학적 샘플을 그대로 건조시키면 샘플이 변형된다. 샘플에 물이 있는 상태에서 동결시키면 전자선의 투과가 어려운 얼음 결정이 생기기 때문에 이 또한 전자현미경 관찰에 어려움을 겪는다.
EMBL의 쟈크 두보쉐(Jacques Dubochet)는 1982년 이러한 문제를 해결할 수 있는 방법을 개발한다. 그는 단백질 샘플을 미세한 망 위에 넣고, 저온의 에탄(Ethane)에 담구어 물을 얇은 필름으로 만들고, 이를 -196도의 액체질소로 냉각하여 샘플주변의 물을 결정형태가 아닌 유리화된 상태로 만들었다. 이러한 처리법을 통하여 단백질 등의 생체 고분자는 주변에 물과 함께 동결된 상태로 유지될 수 있었으며, 동시에 전자선의 투과를 방해하는 얼음의 생성을 막을 수 있었다.[8]
이제 네가티브 염색에서 샘플이 변형되는 등의 부작용 없이 단백질을 전자현미경으로 관찰할 수 있게 되었다. 그러나 여전히 염색 없이 탄소, 수소, 질소로 이루어진 단백질은 전자선을 강하게 산란하지 않았고, 전자현미경에 의해 관찰되는 단백질 입자의 명암 대비는 그리 좋지 않았다. 이를 극복하기 위해서는 또 다른 테크닉이 필요했다.
2차원 화상을 이용한 3차원 입자 재구성
요아힘 프랑크(Joachim Frank)는 1970년대부터 전자현미경으로 관찰된 단백질 분자 영상을 향상시키는 연구를 수행하고 있었다. 전자현미경으로 바라본 단백질 분자는 마치 우리가 해변에 있는 사람을 관찰하는 것처럼 다양한 방향으로 나열되어 있었다. 만약 동일한 방향으로 위치한 분자들이 있다면, 이 분자들의 이미지를 여러 장 모아서 이를 평균을 낸다면, 노이즈가 보정되고 좀 더 선명한 이미지가 나올 것이다. 프랑크는 1980년대 초반 네가티브 염색을 하여 관찰한 여러 종류의 단백질을 이용하여 이것이 가능하다는 것을 입증하였다.
전자현미경으로 관찰된 이미지는 근본적으로 2차원 평면도의 이미지이다. 그러나 전자현미경으로 관찰된 단백질은 다양한 방향으로 분포되어 있었고, 이들을 각각의 방향별로 분류한 다음 이를 나열하여 투사하면, 3차원 이미지를 만들 수 있다. 이러한 테크닉은 처음에는 네거티브 염색으로 얻은 현미경 이미지에 적용되었지만 곧 Cryo-EM에 의해 얻어진 이미지에도 적용되게 되었다. 1990년대 초, 리보솜, 헤모시아닌, 칼슘 방출 채널 등의 다양한 단백질을 Cryo-EM으로 관찰한 결과를 3차원 윤곽으로 만든 구조가 처음으로 제시되었다.[10]
그러나 그 당시의 ‘단백질 윤곽'은 아직 X선 결정학 등으로 얻어낸 원자수준의 모델을 구축할 정도의 고해상도는 아니었고, 단백질의 대략적인 모양을 식별할 정도의 저해상도, 약 9-10옹스트롬 정도의 해상도를 가진 정도의 정보에 그쳤다. 일부 구조생물학자들은 Cryo-EM에서 나온 단백질 윤곽을 충분한 디테일을 식별할 수 없는 ‘덩어리(Blob)'이라고 비하하며 Cryo-EM을 통하여 단백질 구조를 규명해보려는 시도를 ‘블랍볼로지(Blobology)'라고 비하해서 칭하기도 하였다. 실제로 2010년대 초반까지만 하더라도 Cryo-EM에 의한 단백질 구조는 결정구조를 얻을 수 없을때 단백질의 구조에 대한 윤곽만을 알아보기 위한 차선책 정도로 생각되었다.
그러나 이러한 인식은 기술의 발전에 의해서 점점 달라지게 된다.
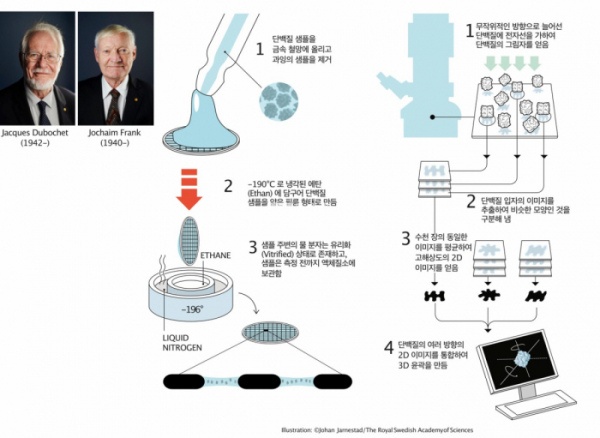
▲그림 1. Cryo-EM을 이용한 단백질 구조결정을 가능케 한 2가지 핵심기술. 자크 두보쉐(Jacques Dubochet)는 단백질 시료를 얇은 망에 넣고 저온 에탄(Ethan)에 넣어서 단백질 샘플을 얇은 필름 상태로 급속 동결시키는 방법을 만들었다. 이렇게하여 전자현미경 관찰을 어렵게 하는 얼음결정이 생기지 않은채 단백질을 현미경으로 관찰할 수 있게 되었다. 요아힘 프랑크(Jochaim Frank)는 전자현미경의 단백질 이미지를 분석하여 이를 비슷한 방향으로 위치한 것끼리 모으고, 동일한 이미지를 평균하여 노이즈를 줄인 고해상도 이미지로 만들고, 이를 이용하여 단백질의 3D 윤곽을 재구성하는 기술을 개발하였다. 두보쉐와 프랑크, 그리고 전자현미경으로 고해상도 단백질 구조를 규명할 수 있다는 것을 처음으로 보여준 리처드 핸더슨(Richard Henderson)은 2017년 Cryo-EM기술 개발의 공로로 노벨화학상을 수상하였다.
해상도 혁명
2010년대 이후 여러가지 기술적 발전에 의해서 저해상도 구조 정보밖에 얻지 못한다는 Cryo-EM의 약점이 점차로 극복되어 이전의 ‘블랍볼로지'라는 인식에서 벗어나 X선 결정학에 비견될 만큼 고해상도의 구조를 얻을 수 있게 되었다. 어떤 기술적인 발전이 여기에 기여하였는가?
첫번째는 전자를 검출하는 검출기(Detector)의 발전이다. 초창기의 전자현미경의 신호는 필름을 이용하여 감광되었으나 그 이후에는 CCD 디텍터 등으로 디지털 이미지 형식으로 전자를 얻는 것으로 발전하였다. CCD 디텍터는 전자를 받아 이를 빛 신호로 변환하고, 이를 검출하는 방식이었다. 그러나 2010년경 등장한 직접 전자 감지 디텍터(Direct electron detector)는 기존의 CCD 디텍터에 비해서 훨씬 더 고감도의 데이터를 얻음으로써, 보다 더 정보량이 높은 데이터를 얻을 수 있었다. 그리고 기존에 비해서 검출속도가 훨씬 더 빨라져서 1초에 수십장의 사진을 찍을 수 있게 되었으므로, 전자선 노출에 의한 시료의 움직임을 보정할 수 있게 됨으로써 노이즈가 훨씬 적은 선명한 영상을 얻을 수 있게 되었다.
두번째는 이미지 검출 및 데이터 프로세싱 기술의 발전이다. 컴퓨터와 영상 처리 기술의 발전은 전자현미경 사진에서 생체고분자 입자를 찾고 분류하는 작업을 자동적으로 수행하게 하였으며, 따라서 더 많은 양의 입자를 분석하여 이를 분류하고 평균화하는 작업을 수행할 수 있게 되었다. 그리고 형성된 3차원 윤곽을 다시 분류하여 서로 다른 형태를 가진 입자로 재분류하고, 이를 이용하여 다시 3차원 윤곽을 형성하는 과정을 통하여 보다 해상력이 높은 3차원 윤곽을 재구성할 수 있었다.
이러한 여러 기술들의 발전에 의해서 Cryo-EM에 의한 구조 정보의 해상력은 급격히 올라갔다. 특히 2010년 중반 이후 Cryo-EM의 해상도 발전은 급격히 향상되어 이전에는 X선 결정학에서나 얻을 수 있던 3옹스트롬 이하의 고해상도 구조 정보를 Cryo-EM을 통해서도 얻을 수 있게 되었다. Cryo-EM 분석에 적절한 특성을 가지고 있어서 Cryo-EM 분석 기술의 벤치마킹에 사용되는 단백질인 아포페리틴(Apoferritin)의 경우 1.2옹스트롬대의 초고해상도 정보도 얻는 것이 가능하다. 이러한 고해상도 구조 정보는 특히 구조기반의 신약개발에서 매우 중요한데, 약물과 단백질과의 결합 방식을 정확히 아는 것이 약물 최적화에 필수적이기 때문이다.
이렇게 Cry에 의해서 원자 수준의 단백질 구조를 결정할 수 있게 되자, 기존에 결정화가 되지 않아서 구조를 결정하지 못했던 수많은 단백질의 구조가 알려지게 되었다. 이들 중 특히 Cryo-EM 에 의한 고해상도 구조 결정이 가능해진 이후 구조가 많이 풀린 부류의 단백질은 여러 개의 단백질로 구성된 단백질 복합체 및 막단백질이다.
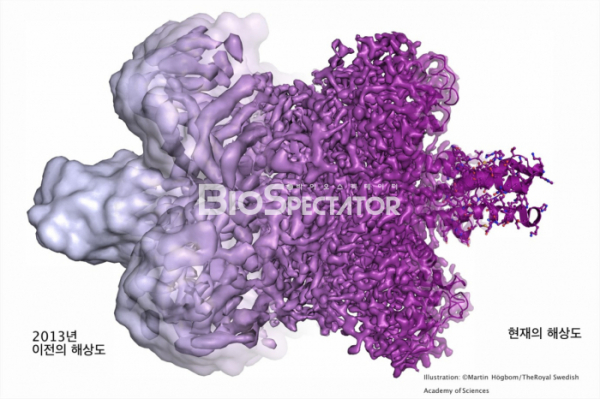
▲그림 2. 2013년 이전의 Cryo-EM에 의한 단백질구조는 낮은 해상도 때문에 단백질의 윤곽만을 식별할 수 있을 정도였고, 일부에서는 충분한 디테일을 식별할 수 없는 ‘덩어리(Blob)'에서 나온 ‘블랍볼로지(Blobology)'로 비하하기도 하였다. 그러나 전자선 디텍터의 향상, 이미지 처리 기술의 향상 등 여러가지 기술적 발전에 의해서 Cryo-EM에의한 단백질 해상도는 향상되었고, 이제는 X선 결정학에 필적할 만큼 원자수준의 고해상도 구조를 풀 수 있게 되었다.
거대 복합체와 막단백질의 구조
생체내의 단백질 중에서는 단독으로 존재하여 기능을 수행하는 것도 있지만, 그것보다는 여러 개의 단백질들이 거대 복합체를 형성하여 기능을 형성하는 것이 더 많다. 가령 자동차와 같은 복잡한 기게에서 개별적인 부품이 조립되어 엔진과 같은 단위 기능을 수행하는 덩어리로 되는 것처럼 우리 몸 속에서 작동하는 많은 생체 반응은 여러 개의 단백질이 결합되어 작용한다.
그러나 기존의 단백질 결정학 기반의 구조생물학 연구에서는 이러한 거대 복합체는 결정을 형성하기 어렵다는 이유 때문에 한정된 복합체들만이 구조가 결정되어 있는 상태였다. 그러나 이제 Cryo-EM의 발전에 의해서 이전에는 제한적인 구조생물학 연구만이 진행되어 오던 이러한 단백질 복합체의 고해상도 구조를 얻을 수 있었다.
이러한 대표적인 예가 진핵생물에서 mRNA가 만들어지는 필수적인 과정인 스플라이싱(Splicing)을 담당하는 스플라이시오솜(Spliceosome)이었다. 스플라이싱이 U1, U2, U3, U4, U5, U6 으로 약칭되는 RNA-단백빌 복합체인 snRNP에 의해서 이루어진다는 것은 1970년대 중반부터 연구되어 왔으나, 이들의 구조에 대한 정보가 미흡했기때문에 정확한 작동기전이 알려지지 않은 상태였다. 2000년대 초반, 스플라이시오솜의 여러 상태에 대한 Cryo-EM 분석이 최초로 시도되었으나, 이들은 약 10옹스트롬 이하의 저해상도 구조여서 그 작동기전에 대한 정확한 힌트를 줄 수 없었다. 2015년 중국 칭화대의 이공 시(Yigong Shi) 연구그룹은 4개의 RNA와 37개의 단백질로 구성된 분자량 1.2메가달톤에 달하는 효모의 스플라이시오솜 구조를 3.8 옹스트롬 해상도로 최초로 규명하였다.[11] 최초의 스플라이시오솜 구조는 비활성화된 구조였지만 이후 여러 단계의 스플라이솜 구조가 밝혀졌다. 2017년에는 인간의 스플라이시오솜 구조 역시 밝혀졌다.
생명 현상에서 중요한 역할을 하는 거대 단백질 복합체 중에서 Cryo-EM의 발전에 의해서 구조가 처음 밝혀진 것 중의 하나는 미토콘드리아 호흡 복합체(Respiratory complex I)이다. 세포 내의 미토콘드리아 내에서 물질을 분해하여 에너지를 형성하기 위해서는 물질 분해에서 나온 전자를 전달받아 이를 미토콘드리아 내막 내의 수소 이온 형태의 전기 에너지로 변환시켜야 한다. 이러한 과정을 수행하는 40개가 넘는 단백질로 구성된 거대 복합체가 호흡 복합체 I 인데, 세균 유래의 호흡 복합체 I 의 구조는 2000년대 중반 X선 결정학에 의해서 구조가 밝혀졌지만, 진핵생물의 미토콘드리아에 있는 호흡 복합체 I 의 구조는 알려지지 않았다. 2014년 소 미토콘드리아의 호흡 복합체의 구조가 약 5옹스트롬 정도의 해상도로 최초로 규명되었고[12], 기술의 발전에 의해서 2020년에는 2.5옹스트롬 수준의 고해상도 구조가 밝혀져서, 전자가 전달되고 이에 따라 수소 이온이 생체막을 가로지르는 과정에서 일어나는 아미노산 수준에서의 구조적인 변화 과정을 알 수 있게 되었다.[13]
Cryo-EM의 발전은 단백질 결정학에서 쉽게 규명되지 않던 주된 단백질인 막단백질의 구조 규명에서 획기적인 발전을 이룩했다. 이전의 연재에서 설명한 것처럼 결정학을 통하여 몇 종류의 막 단백질의 구조가 알려지긴 했지만, 아직 알려지지 않은 막단백질이 훨씬 더 많던 상황에서 Cryo-EM은 막단백질의 구조를 푸는 주된 테크닉이 되었다. Cryo-EM에 의해서 구조가 규명된 생물학적으로 중요한 막단백질만 하더라도 무수히 많은데, 감마-시크리테이즈(gamma-secretase, 2015년), 낭포성 섬유증 막 전도조절자(Cystic Fibrosis Transmembrane Regulator,2017년), 캡사이신 수용체(TRPV1, 2013년), T세포 수용체-CD3 복합체(2019년) 등과 같은 많은 막단백질의 구조가 규명되었다.[14-17]
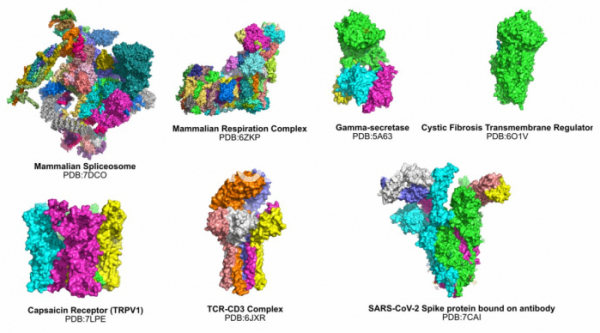
▲그림 3. Cryo-EM에의해 규명된 단백질 복합체/막단백질의 일부. 기존의 단백질 결정학으로는 불가능했던 수많은 거대 복합체와 막단백질의 구조가 Cryo-EM을 통하여 속속 밝혀지고 있다.
Cryo-EM과 의약품 개발
그동안 결정화가 안되어 고해상도 구조 정보를 얻을 수 없던 많은 단백질들의 구조가 Cryo-EM의 발전에 의해서 하나둘씩 규명된 이후, 많은 사람들은 Cryo-EM에 의한 구조분석이 신약개발에 어떤 도움을 줄 것인지를 고민하기 시작하였다.
현재까지 개발된 많은 약물의 타깃 중 상당수가 막단백질이고, 기존에는 결정화 문제 때문에 구조 정보를 얻을 수 없던 상당수의 막 단백질의 구조가 Cryo-EM에 의해 규명된 이후, Cryo-EM에 의한 구조기반 소분자 약물개발을 하려는 노력이 진행되고 있다. 특히 현재까지 FDA에서 승인된 약물 중 약 35% 정도가 GPCR을 표적으로 하고 있으며, 약 128종의 GPCR이 약물 표적으로 알려져 있다는 것을 생각한다면, Cryo-EM 기반의 약물 개발에서 각종 GPCR이 가장 우선적인 타깃으로 되는 것은 놀라운 일이 아니다.
GPCR기반의 소분자 약물개발에서 한가지 극복되어야 할 점이 있다면, 통상적으로 X선 결정학으로 약물을 개발할 때는 1옹스트롬 대에 달하는 원자 수준의 고해상도 구조가 필요하지만, Cryo-EM을 통한 구조결정으로는 주요 약물 타겟에 대해서 그정도의 고해상도 구조 정보를 얻어내는 것은 쉬운 일이 아니다. 물론 이를 극복하기 위한 노력이 지속적으로 진행되고 있으며, 최근의 연구 결과에 따르면 약물과 단백질간의 상호작용을 파악하기에 충분한 2.5Å 이하 해상도의 고해상도 구조를 얻어내는데 성공하였으므로, GPCR을 비롯한 여러가지 막단백질 타겟을 대상으로 한 구조기반 약물개발도 본격적으로 진행될 것으로 보인다.
항체 신약 분야에서도 Cryo-EM은 중요한 툴로 사용되고 있다. 항체와 항원 복합체의 경우 결정화되어 고해상도 구조를 얻은 경우가 있긴 하지만, 역시 다른 단백질과 마찬가지로 모든 항체-항원 복합체를 결정화하여 구조를 얻는다는 것은 불가능에 가깝다. 이러한 상황에서 결정화 없이 구조를 얻을 수 있는 Cryo-EM은 매우 중요한 역할을 한다. 특히 아직 항체와 결합하는 항원의 에피토프, 항원에 결합하는 항체의 파라토프가 알려져 있지 않은 상황에서 Cryo-EM을 이용한 항원-항체 복합체 구조의 결정은 신속하게 에피토프/파라토프의 정보를 알 수 있는 방법이 되었다. 최근 코로나19 판데믹에서도 Cryo-EM에 의한 구조규명은 그 위력을 발휘하였다. 코로나19의 원인 바이러스인 SARS-CoV-2의 표면에 존재하는 스파이크 단백질은 바이러스가 세포에 침투하는데 필수적인 역할을 한다. 코로나19 판데믹이 시작된지 불과 1개월이 되지 않은 상황에서 스파이크 단백질의 구조가 Cryo-EM으로 규명되었고, 이들의 구조가 어떻게 변화하는지가 알려졌다. 그리고 스파이크 단백질과 세포의 SARS-CoV-2 수용체인 ACE2와의 결합 방식, 그리고 스파이크 단백질과 결합하여 바이러스의 감염을 막아주는 중화항체들이 각각 어떻게 결합하는지가 Cryo-EM을 통한 구조분석을 통하여 신속하게 알려졌다. 이렇게 Cryo-EM에 의한 구조결정 기술은 신약개발에도 점점 널리 활용될 것으로 보인다.
지금까지 알아본 것처럼 Cryo-EM 기술은 이제 실험구조생물학에 있어서 필수적인 기술로써, 그동안 X선 결정학에 의해 규명되기 힘들었던 거대 복합체나 막단백질 등에 대한 구조 정보를 얻어내는데 활발하게 사용되고 있다. 어떻게 보면 1950년대 이후 구조생물학의 중심된 기술이었던 X선 결정학의 뒤를 이은 주류 연구방법이 되었다고 볼 수 있을 것이다. 물론 Cryo-EM에 의해서 구조를 얻는그러나 어쨌든 이전에는 결정화가 되지 않아서 아예 구조 정보를 얻을 수 없을 것으로 생각되었던 수많은 단백질들의 구조를 얻을 수 있는 방법이 출현한 것은 의미가 크다. 특히 최근의 알파폴드와 같은 단백질 구조 예측 기술은 Cryo-EM에 의한 거대 복합체의 구조 정보 획득과 함께 결합되면 구조결정의 효율을 훨씬 더 올려줄 것이다. 현재의 구조예측 기술은 단일 단백질 가닥에 대해서는 상당히 정확한 예측이 가능하지만, 단백질 복합체의 구조 예측은 아직도 쉬운 문제가 아니기 때문이다.
참고문헌
1. 현재경. (2017). 초저온 전자현미경법을 통한 고분해능 생물분자 구조분석. 진공이야기 , 4(4), 18-22.
2. Booth, David S., Agustin Avila-Sakar, and Yifan Cheng. "Visualizing proteins and macromolecular complexes by negative stain EM: from grid preparation to image acquisition." JoVE (Journal of Visualized Experiments) 58 (2011): e3227.
3. Lake, J. A. (1976). Ribosome structure determined by electron microscopy of Escherichia coli small subunits, large subunits and monomeric ribosomes. Journal of molecular biology , 105(1), 131-159.
4. Kirschner, M. W., Honig, L. S., & Williams, R. C. (1975). Quantitative electron microscopy of microtubule assembly in vitro. Journal of molecular biology, 99(2), 263-276.
5. Tanaka, K., Ii, K., Ichihara, A., Waxman, L., & Goldberg, A. L. (1986). A high molecular weight protease in the cytosol of rat liver. I. Purification, enzymological properties, and tissue distribution. Journal of Biological Chemistry, 261(32), 15197-15203.
6. Henderson, R., & Unwin, P. N. T. (1975). Three-dimensional model of purple membrane obtained by electron microscopy. Nature, 257(5521), 28-32.
7. Henderson, R., Baldwin, J. M., Ceska, T. A., Zemlin, F., Beckmann, E., & Downing, K. H. (1990). Model for the structure of bacteriorhodopsin based on high-resolution electron cryo-microscopy. Journal of molecular biology, 213(4), 899-929.
8. Dubochet, J., Adrian, M., Lepault, J., and McDowall, A. W. (1985) Emerging techniques: Cryo-electron microscopy of vitrified biological specimens. Trends Biochem. Sci. 10, 143-146
9. Radermacher, M., Wagenknecht, T., Verschoor, A., and Frank, J. (1986) A new 3D reconstruction scheme applied to the 50s ribosomal subunit of E. coli. J. Microsc. 141, RP1-RP2; Radermacher,M.,Wagenknecht,T.,Verschoor,A.,and Frank,J.(1987)Three-dimensional reconstruction from a single-exposure, random conical tilt series applied to the 50S ribosomal subunit of Escherichia coli. J. Microsc. 146, 113-136
10. Lambert, O., Boisset, N., Penczek, P., Lamy, J., Taveau, J. C., Frank, J., & Lamy, J. N. (1994). Quaternary structure of Octopus vulgaris hemocyanin: three-dimensional reconstruction from frozen-hydrated specimens and intramolecular location of functional units Ove and Ovb. Journal of molecular biology, 238(1), 75-87; Radermacher, M., Rao, V., Grassucci, R., Frank, J., Timerman, A. P., Fleischer, S., & Wagenknecht, T. (1994). Cryo-electron microscopy and three-dimensional reconstruction of the calcium release channel/ryanodine receptor from skeletal muscle. The Journal of cell biology, 127(2), 411-423.
11. Yan, C., Hang, J., Wan, R., Huang, M., Wong, C. C., & Shi, Y. (2015). Structure of a yeast spliceosome at 3.6-angstrom resolution. Science, 349(6253), 1182-1191.
12. Vinothkumar, K. R., Zhu, J., & Hirst, J. (2014). Architecture of mammalian respiratory complex I. Nature, 515(7525), 80-84.
13. Kampjut, D., & Sazanov, L. A. (2020). The coupling mechanism of mammalian respiratory complex I. Science, 370(6516), eabc4209.
14. Bai, X. C., Yan, C., Yang, G., Lu, P., Ma, D., Sun, L., ... & Shi, Y. (2015). An atomic structure of human γ-secretase. Nature, 525(7568), 212-217.
15. Liu, F., Zhang, Z., Csanády, L., Gadsby, D. C., & Chen, J. (2017). Molecular structure of the human CFTR ion channel. Cell, 169(1), 85-95.
16. Liao, M., Cao, E., Julius, D., & Cheng, Y. (2013). Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature, 504(7478), 107-112.
17. Dong, D., Zheng, L., Lin, J., Zhang, B., Zhu, Y., Li, N., ... & Huang, Z. (2019). Structural basis of assembly of the human T cell receptor–CD3 complex. Nature, 573(7775), 546-552.





![[인사]한미그룹, 임원 승진 인사](https://img.etoday.co.kr/crop/74/74/2071974.jpg)








![[인사]한미그룹, 임원 승진 인사](https://img.etoday.co.kr/crop/77/77/2071974.jpg)



