오피니언
[남궁석의 신약연구史]아스피린 타깃 'COX' 알기까지
남궁석 SLMS(Secret Lab of Mad Scientist) 대표
[염증과의 싸움②]프로스타글란딘 경로의 발견과 비스테로이드 소염제의 등장
지난 연재에서는 천연물 유래의 살리실산과 ‘아스피린(아세틸살리실산)'이 최초의 진통 소염제로 등장하는 과정을 알아보았다. 아스피린은 20세기 초부터 가장 널리 사용되는 약물이 되었지만 과연 어떤 기전으로 아스피린의 진통 소염 작용이 일어나는지를 아는데는 20세기 후반이 되어야만 했다. 이번 연재에서는 아스피린의 작용 기전을 이해하기 위하여 어떤 발견이 수반되어야 했는지를 알아보며, 아스피린을 대체할 새로운 비스테로이드성 소염제가 등장하는 과정을 알아보고자 한다.
프로스타글란딘(Prostaglandin)
우리가 신약연구사에서 알아본 새로운 약물을 탄생하게 한 기전 연구 등의 상당수는 원래는 해당 연구의 목적과는 상이한 연구에서부터 비롯되는 경우가 많으며 아스피린 등의 비스테로이드성 소염제가 작용하는 대사인 프로스타글란딘(Prostaglandin) 및 이의 합성 경로 역시 그 시초는 소염제나 염증 연구와는 별 관련없는 연구로부터 비롯되었다.
19세기 말에서 20세기 초반은 에피네프린(Epinephrine) 등의 호르몬들이 발견되고 이들이 어떻게 여러가지 생리활성을 조절하는지에 대한 연구가 활발히 수행되던 시기이다. 프로스타글란딘의 발견 역시 이러한 시대적 상황과 연관되어 있다.
1930년 미국의 산부인과 의사인 커즈로크(Raphael Kurzrok)와 리엡(Charles C. Lieb)이 인공수정 도중에 정액에 자궁 수축을 유도하는 성질이 있으며, 이것은 정자에 의해서 유도되는 것이 아닌 정액 내에 있는 물질에 의해서 유도된다는 것을 밝혔다[1]. 이후 1935년 스웨덴의 생리학자 울프 본 오일러(Ulf Von Euler, 1905-1983)에 의해 해당 물질이 유래된다고 생각한 전립선(Prostate gland)을 따서 프로스타글란딘(Prostaglandin)이라고 명명하였다[2].
프로스타글란딘은 애초에 전립선에서 발견되었지만, 그 이후 프로스타글란딘의 합성은 전립선 뿐만 아니라 거의 모든 조직과 세포에서 이루어진다는 것이 알려지게 되었다. 오일러는 프로스타글란딘이 혈관 확장과 근육 수축을 유도한다는 결과를 얻었지만 프로스타글란딘의 정확한 화학 구조와 프로스타글란딘이 어떤 과정을 거쳐서 생합성되는지에 대해서는 알아내지 못했다[3]. 프로스타글란딘의 생리적 기능을 보다 정확히 알기 위해서는 순수한 프로스타글란딘을 정제하여 이들의 생리 효과를 정확히 파악해야만 했기 때문이다. 그러나 2차 세계대전이 발발한 이후 프로스타글란딘에 대한 연구는 크게 진척이 없었다.
스웨덴의 수네 뵈리스트룀(Sune Karl Bergström, 1916-2004)은 지방산의 산화 과정을 연구하던 생화학자였다. 그는 1945년 학회에서 프로스타글란딘을 처음 발견한 오일러를 만나게 된다. 오일러는 양의 정소에서 추출한 지방 추출액에 프로스타글란딘이 많이 들어 있다는 것은 알고 있었지만, 여기서 생물학적 활성을 가지는 물질만을 추출하지는 못하고 있었다. 오일러는 지방산 연구를 한 뵈리스트룀과 협력 연구를 제안하였고, 그가 전쟁 전부터 모은 정소 유래 지방 추출액을 뵈리스트룀에게 제공하였다. 뵈리스트룀은 1948년 룬드 대학에 임용된 이후 대학원생 벵트 사무엘슨(Bengt Ingemar Samuelsson, 1934-)과 함께 프로스타글란딘의 정제와 화학구조 결정 연구를 시도하기 시작한다[4].
이들은 1957년 스웨덴과 노르웨이 양의 정소 추출액에서 향류추출(counter current extraction)과 분배 크로마토그래피(portion chromatography) 등의 테그닉을 이용하여 생리 활성을 가지는 프로스타글란딘 E와 프로스타글란딘 F를 정제하고[5]. 이들의 화학 구조를 1962-1963년 결정하였다[6].
프로스타글란딘(Prostaglandin)의 생합성 경로 규명
뵈리스트룀과 함께 프로스타글란딘의 화학구조를 결정하는데 공헌했던 벵트 사무엘슨은 동위원소로 표지된 탄소 20개짜리 지방산인 아라키돈산(arachidonic acid)이 정소 추출액에 의해서 프로스타글란딘 H로 전환되는 것을 발견하였고, 이 과정에서 중요한 역할을 하는 효소인 사이클로옥시게나아제(Cyclooxygenase)를 발견하였다. 이렇게 생성된 프로스타글란딘 H는 다양한 프로스타글란딘의 형태로 변환되게 되고, 이들은 종류에 따라서 다양한 생물학적 활성을 가진다. 가령 프로스타글란딘 E2(PGE2)는 평활근의 수축, 말초 혈관 확장, 발열 및 통증 전달 작용 등 염증 작용에 관련된 수많은 반응을 매개하게 된다[7].
그렇다면 아라키돈산은 어떻게 만들어지는가? 아라키돈산은 세포막의 인지질(Phospholipids)로부터 포스포리파아제 A2(PLA2)에 의해 만들어진다. 포스포리파아제 A2는 인지질의 트리글리세롤에 붙어있는 3개의 지방산 중 두번째 위치의 지방산을 분해하여 아라키돈산과 라이소포스파티딕산(Lysophosphatidic acid)으로 전환한다. 포스포리파아제 A2는 외부 상처나 감염 등에 의해서 활성화되어 세포막의 인지질로부터 아라키돈산을 형성하고, 이렇게 형성된 아라키돈산은 프로스타글란딘 E(PGE)과 트롬복산(Thromboxane), 프로스타사이클린(Prostacyclin) 등의 산물로 변화하여 다양한 생리 활성을 나타낸다.
여기서 염증 반응과의 관계가 떠오르게 되는데 세포가 물리적 손상이나 병원균의 침입 등 다양한 원인으로 손상되면 세포 밖으로 포스포리파아제 A2가 방출되고, 방출된 포스포리파아제 A2 는 인지질로부터 아라키돈산을 만들어 내고, 아라키돈산의 농도 증가는 프로스타글란딘의 합성 증가를 매개하여 염증 반응을 매개하게 되는 것이다[8].

▲그림1: 염증 반응의 주된 신호전달분자인 프로스타글란딘 (Prostaglandin)의 연구 주역과 그 합성경로. (상) 수네 뵈리스트룀 (Sune Karl Bergström, 1916-2004) 은 양의 정소에서 프로스타글란딘을 정제하고 그 화학구조를 규명하였으며 벵트 사무엘슨 (Bengt Ingemar Samuelsson, 1934-)은 프로스타글란딘의 전체 합성 및 대사 경로를 규명하였다. 존 베인 (John R Vane, 1927-2004)은 아스피린 등의 비 스테로이드성 소염진통제가 프로스타글란딘 합성 경로에서 아라키돈산을 프로스타글란딘 H2 로 전환시키는 사이클로옥시게나아제 (Cyclooxygenase, COX)를 저해함으로써 프로스타글란딘 합성을 저해하여 효과를 낸다는 것을 규명하였다.
60년만에 발견된 아스피린의 작용 기전
이렇게 프로스타글란딘의 생합성 경로가 밝혀지게 되는 1960년대 이후, 이러한 연구에 기반하여 기존에 작용기전을 모른채 소염, 해열 작용 등으로 사용되고 있던 아스피린의 작용 기전을 규명하고자 하는 연구가 시작되었다. 특히 아스피린은 이전부터 위궤양 출혈 등을 유발한다는 것이 잘 알려져 있었는데[9], 1968년 아스피린이 혈소판 기능을 저해한다는 것이 밝혀지면서[10] 이러한 아스피린의 약리적 작용이 어떻게 일어나는지에 대한 관심이 높아지기 시작하였다.
1971년 영국의 약리학자 존 베인(John R Vane, 1927-2004)는 아스피린의 약리적 작용이 프로스타글란딘의 합성과 관련이 있을 것이라는 가설하에 이를 입증하려는 실험을 했다. 그는 기나아 피그의 폐 세포 추출물을 이용하여 시험관에서 아라키돈산으로부터 프로스타글란딘을 합성하는 효소 반응을 실시하였는데, 여기서 아스피린과 소디움 살리실산, 그리고 1963년에 등장한 새로운 소염제인 인도메타신(indomethacin)을 첨가해 보았다[11]. 이 3가지 모두 농도 의존적으로 프로스타글란딘 생합성을 억제하는 것을 보였다. 또한 개에서 적출한 비장(Spleen)을 이용한 실험에서 인도메타신과 아스피린 처리에 의해서 비장에서의 프로스타글란딘 방출을 억제한다는 것을 입증하였다[12]. 이것으로써 아스피린과 새로 등장한 비스테로이드성 소염제가 모두 유사하게 프로스타글란딘을 억제하는 것을 알게 되었다. 그렇다면 아스피린 등이 억제하는 프로스타글란딘의 합성 단계는 어디일까? 1976년 양의 정소로부터 사이클로옥시게나아제(COX)의 정제가 이루어졌고 1988년에 유전자가 발견되었고, 1994년 단백질 입체 구조가 규명되었다[13]. 이러한 정보들은 아스피린이 어떻게 작용하는지에 대한 원자 수준의 기작을 알려주게 된다. 1982년 수네 뵈리스트룀, 벵트 사무엘슨, 존 베인은 프로스타글란딘의 정제 및 합성경로 규명, 그리고 아스피린의 약리 기전 발견의 공로로 노벨 생리의학상을 공동수상한다.
COX
COX는 막에 결합된 분자량 7만2,000달톤(Da)의 단백질로써 아라키돈산을 산화시켜 프로스타글란딘 G2(Prostaglandin G2)를 형성한다. 아스피린은 이 과정에서 효소 작용에 필수적인 530번째 세린과 반응하여 아스피린의 아세틸기는 세린기로 전이된다. 아세틸화된 세린 때문에 효소 반응이 일어나는 활성자리는 좁아지고, 기질인 아라키돈산은 더이상 활성자리에 들어오지 못하기 때문에 효소 반응은 억제된다. 즉 아스피린은 비가역적으로 COX의 활성을 억제하며, 이는 아스피린(아세틸살리신산) 이전에 소염 작용이 알려졌던 살리실산과는 다른 기전이다. 즉 살리실산은 아스피린(아세틸살리실산)과 마찬가지로 COX의 활성자리에 결합하지만, 살리실산에는 아세틸기가 없기 때문에 COX에서 떨어질 수 있으며, 따라서 가역적인 경쟁적 저해제(Reversible competitive inhibitor)로 작용한다.
이렇게 아스피린의 진통, 해열작용과 같은 약리적 작용 및 위장에서의 출혈, 혈소판 응집 억제 등과 같은 부작용 등은 COX를 비가역적으로 억제하여 일어나는 것이라는 것이 아스피린이 발견된 이후 약 80년이 넘어서야 알려지게 되었다. 그러나 아스피린 이후에 등장한 많은 비 스테로이드성 소염제는 서로 다른 특성을 가지고 있고, 아스피린은 진통, 해열작용이 좋은 반면 항염증 작용에는 훨씬 더 많은 용량이 필요한데, 다른 비 스테로이드성 소염제는 아스피린보다 훨씬 더 좋은 항염증 작용을 보였다. 이러한 차이가 나타나는 이유는 무엇일까?
이러한 의문은 1991년에 또 다른 COX 가 존재한다는 것이 알려지면서 풀리게 되었다. 제일 먼저 발견된 COX(COX-1으로 다시 이름지어진)의 경우 항상 발현되는(Constitutive) 성질을 가지고 있는 반면 COX-2의 경우에는 박테리아 감염시나 성장인자의 자극 등 외부적인 염증 자극이 있을때만 발현된다.
COX-1 과 COX-2는 아미노산 서열 기준으로 약 71% 정도의 상동성을 가지고 있다. 두 효소는 동일한 효소 작용을 촉매하므로 효소 작용이 일어나는 활성자리도 거의 비슷하지만 약간의 차이가 존재한다. COX-2의 경우에도 아스피린이 작용하여 아세틸화하는 세린이 존재하지만, COX-2의 활성자리는 COX-1에 비해서 조금 더 여유가 있다. 즉 COX-1에 이소류신이 있는 위치에 COX-2 에는 조금 더 크기가 작은 아미노산인 발린이 존재하므로 활성자리의 공간이 좀 더 넓은 편이다. 따라서 아스피린에 의해서 아세틸화가 일어나면 아라키돈산이 전혀 결합을 할 수 없는 COX-1에 비해서 COX-2에서는 아스피린에 의해서 아세틸화가 일어나도 아라키돈산이 그 틈을 비집고 들어가는 것이 가능하다[14].
아스피린에 의해서 아세틸화된 COX-2에서는 아라키돈산은 프로스타글란딘 G2로 촉매되지 못하고 15-R-히드록시에이코사테트라노익산이라는 물질이 만들어진다. 이 물질 역시 항염증 작용을 하지만, 그 항염증 작용은 COX-2의 활성을 완전히 억제하는 다른 비 스테로이드 소염제와는 조금 다른 기전인 셈이다.
비스테로이드성 소염제(NSAIDS)
한편 아스피린의 타겟이 COX라는 것이 알려지기 전부터 아스피린의 위 출혈 유발 등의 부작용이 알려졌고, 이러한 부작용을 줄이고 좀 더 효과가 있는 ‘아스피린의 대체 약품’을 찾기 위한 연구가 1960년대에 활발히 진행되었다.
이렇게 등장한 ‘아스피린의 대체품’ 중의 하나가 이부프로펜(Ibuprofen)이다. 이부프로펜은 영국의 제약사 부츠(Boots)의 화학자 스튜어트 애덤스(Steward Adams, 1923-2019)에 의해서 개발되었다. 애덤스가 이끄는 연구팀은 아스피린의 진통 억제 능력은 그대로 유지한채 위출혈 등의 부작용이 줄어든 류마치스성 관절염 환자에게 투여할 약물을 찾고 있었다. 이들은 페닐프로파노익산(Phenylpropanoic acid)의 유도체를 만들었고, 이 중에서 가장 효과가 좋은 2-(4-이소부틸페닐) 프로파노익산이 선택되었다. 1961년에 처음 합성된 이 약물은 동물 및 인간 대상 임상시험을 통하여 같은 용량의 아스피린에 비해서 1/3의 용량으로도 좋은 효능을 보였다. 후에 COX가 발견된 이후 COX에 대한 저해력을 아스피린과 비교해 보니, 이부프로펜은 아스피린에 비해 4배, 그리고 COX-2의 활성을 거의 저해하지 못하는 아스피린에 비해서 이부프로펜은 COX-2 역시 저해하는 효과를 가진다는 것이 밝혀졌다. 이부프로펜은 영국에서 1969년, 미국에서는 1974년 사용되기 시작하였으며, 이부프로펜은 1983년부터 미국과 영국에서 의사의 처방 없이 구매할 수 있는 일반의약품(Over the Counter Medicine)으로 판매되기 시작하였다.
그렇다면 같은 타겟인 COX를 저해하는 아스피린과 이부프로펜은 어떤 차이가 있을까? 활성자리의 세린에 아세틸기를 옮겨셔 효소를 비가역적으로 저해하는 아스피린에 비해서 이부프로펜은 아세틸기가 없으므로 효소를 비가역적으로 저해하지는 못하지만, 효소의 결합자리에 강하게 결합하는 경쟁적 저해제로써 역할한다. 또한 아스피린은 염증 반응에서 주로 역할하는 COX-2 를 직접적으로 저해하지 못하지만 이부프로펜은 COX-2 역시 COX-1과 마찬가지로 저해하여 프로스타글란딘 G2 의 생성을 억제한다는 차이가 있다.
앞에서 설명한 것처럼 프로스타글란딘은 몸 속에서 수많은 생명현상에 관여하며, 여기에는 염증 반응 위에에도 위산 분비 억제작용, 위점막 보호 작용 등이 존재한다.
아스피린, 이부프로펜 등의 대부분의 비 스테로이드성 소염제는 COX-1과 COX-2 를 모두 비선택적으로 저해한다. 그러나 프로스타글란딘에 의한 위산 분비 억제작용, 위점막 보호작용은 주로 COX-1에 의해서 수행되며, 아스피린 등에 의해서 COX-1이 억제되는 것은 아스피린의 가장 큰 부작용인 위출혈을 일으키는 원인이 된다.
그렇다면 이러한 COX-1을 저해함으로써 생기는 부작용 없이 COX-2만을 특이적으로 저해한다면 위출혈과 같은 기존의 비 스테로이드 소염제의 부작용이 없이 염증 작용만 저해할 수 있는 약물을 만들 수 있지 않을까? 이러한 가설에 기반하여 COX-2가 발견되고 이의 기능이 밝혀진 1990년대 이후 제약사들은 COX-2를 선택적으로 저해하는 차세대 비스테로이드성 소염제의 개발에 나섰다.
다음 연재에서는 COX-2를 선택적으로 저해하는 비스테로이드성 소염제의 개발과 예상치 못한 새로운 부작용 및 이에 따른 리콜 과정에 대해서 알아보도록 한다.
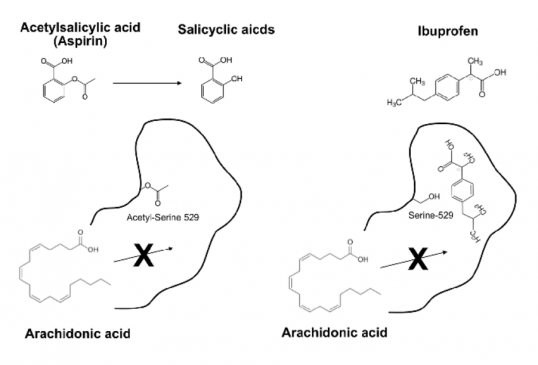
▲그림2: 대표적인 비 스테로이드계 소염진통제인 아스피린과 이부프로펜의 기전 차이. 아스피린은 사이클로옥시게나아제 (COX)의 활성자리에 있는 529번째 세린을 아세틸화시켜 아세틸세린으로 만든다. 기질인 아라키돈산은 세린에 아세틸기가 붙어서 좁아진 활성자리에 들어가지 못하기 때문에 반응이 억제되며, 사이클로옥시게나아제는 비가역적으로 불활성화된다. 반면 이부프로펜은 세린과 반응하지는 않지만 아라키돈산이 결합할 결합자리에 결합하여 기질이 들어가지 못하도록 만든다. COX-2 의 경우 기질의 결합자리가 COX-1 에 비해서 비교적 넓기 때문에 아스피린과 세린이 반응해도 아라키돈산이 반응할 수 있으며, 이때는 원래 효소의 산물인 프로스타글란딘 H2 대신15-R-히드록시에이코사테트라노익산이라는 물질이 만들어져 항염증 작용을 한다. 반면 이부프로펜은 COX-1과 COX-2 에 모두 작용하여 프로스타글란딘 H2의 형성을 막게 된다.
참고문헌
Kurzrok, R., & Lieb, C. C. (1930). Biochemical Studies of Human Semen. II. The Action of Semen on the Human Uterus. Experimental Biology and Medicine, 28(3), 268–272. doi:10.3181/00379727-28-5265
Euler, U. S. (1935). Über die Spezifische Blutdrucksenkende Substanz des Menschlichen Prostata- und Samenblasensekretes. Klinische Wochenschrift, 14(33), 1182–1183. doi:10.1007/bf01778029
Von Euler, U. S. (1936). On the specific vasodilating and plain muscle stimulating substances from accessory genital glands in man and certain animals (prostaglandin and vesiglandin). The Journal of physiology, 88(2), 213-234.
Explorable.com (May 6, 2011). Prostaglandins and Biologically Active Substances. Retrieved Jan 07, 2020 from Explorable.com: https://explorable.com/prostaglandins; Bergström, S. (1983). The prostaglandins: from the laboratory to the clinic (Nobel lecture). Angewandte Chemie International Edition in English, 22(11), 858-866.
Bergström, S., & Sjövall, J. (1960). The isolation of prostaglandin F from sheep prostate glands. Acta chem. scand, 14(8).
Bergstrom, S., Ryhage, R., Samuelsson, B., & Sjovall, J. (1962). The structure of prostaglandin E F1 and F2. Acta Chemica Scandinavica, 16(2), 501-2.
Samuelsson, B. E. N. G. T., Goldyne, M., Granström, E., Hamberg, M., Hammarström, S., & Malmsten, C. (1978). Prostaglandins and thromboxanes. Annual review of biochemistry, 47(1), 997-1029.
Dennis, E. A., Cao, J., Hsu, Y.-H., Magrioti, V., & Kokotos, G. (2011). Phospholipase A2 Enzymes: Physical Structure, Biological Function, Disease Implication, Chemical Inhibition, and Therapeutic Intervention. Chemical Reviews, 111(10), 6130–6185.doi:10.1021/cr200085w
Douthwaite AH, Lintott GM: Gastroscopic observation of the effect of aspirin and certain other substances on the stomach. Lancet, 1935, ii, 1222–1225.
O’Brien JR: Effects of salicylates on human platelets. Lancet, 1968, i,779.
VANE, J. R. (1971). Inhibition of Prostaglandin Synthesis as a Mechanism of Action for Aspirin-like Drugs. Nature New Biology, 231(25), 232–235. doi:10.1038/newbio231232a0
Ferreira; Moncada, S; Vane, JR (1971). "Indomethacin and aspirin abolish prostaglandin release from the spleen". Nature New Biology. 231 (25): 237–9.
Hemler, M. A. R. T. I. N., & Lands, W. E. (1976). Purification of the cyclooxygenase that forms prostaglandins. Demonstration of two forms of iron in the holoenzyme. Journal of Biological Chemistry, 251(18), 5575-5579; DeWitt DL, Smith WL. Primary structure of prostaglandin G/H synthase from sheep vesicular gland determined from the complementary DNA sequence. Proc Natl Acad Sci USA. 1988;85:1412–1416.; Picot, D., Loll, P. J., & Garavito, R. M. (1994). The X-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature, 367(6460), 243.
Botting, R. M. (2010). Vane’s discovery of the mechanism of action of aspirin changed our understanding of its clinical pharmacology. Pharmacological Reports, 62(3), 518-525.
3.















